Naukowcy rozszyfrowali mechanizm molekularny, w którym receptor na powierzchni komórki należący do rodziny enzymów wiążących czynniki wzrostu, regulujący różnicowanie, proliferację, przeżycie, metabolizm i migrację komórek, zapobiega nowotworom.
Enzym ten zwany VEGFR1 wstrzymuje ekspresję siebie (jest autoinhibitowany) w przypadku braku liganda – na przykład hormonów. Badania mogą wskazać drogę do opracowania rozwiązań medycznych w leczeniu raka okrężnicy i nerek poprzez wykorzystanie cząsteczek, które preferencyjnie stabilizują nieaktywny stan VEGFR1.
Receptory na powierzchni komórki, takie jak receptorowe kinazy tyrozynowe (RTK), odgrywają kluczową rolę w przekształcaniu sygnałów zewnątrzkomórkowych (od sygnałów chemicznych, takich jak czynniki wzrostu, ogólnie określane jako ligandy), w ściśle regulowaną odpowiedź komórkową. Wiązanie liganda z receptorami zewnątrzkomórkowymi aktywuje enzymy sprzężone wewnątrzkomórkowo (kinazy tyrozynowe). Aktywowany enzym z kolei dodaje grupę fosforanową do kilku cząsteczek tyrozyny, które działają jako adapter do składania kompleksu sygnalizacyjnego. Tworzenie kompleksu sygnalizacyjnego reguluje różnorodne funkcje komórkowe, takie jak wzrost i rozwój komórek oraz odpowiedź immunologiczna gospodarza. Spontaniczna aktywacja RTK przy braku ligandów jest często powiązana z wieloma ludzkimi patologiami, takimi jak nowotwory, cukrzyca i choroby autoimmunologiczne. Naukowcy badają, w jaki sposób komórka utrzymuje stan autoinhibicji enzymu i dlaczego taka autoinhibicja zostaje naruszona w trakcie postępu patologii u człowieka.
Naukowcy z Indyjskiego Instytutu Edukacji Naukowej i Badań Naukowych (IISER) w Kalkucie zbadali jeden z takich RTK, zwany receptorem czynnika wzrostu śródbłonka naczyniowego (VEGFR). Rodzina receptorów VEGFR jest kluczowym regulatorem procesu powstawania nowych naczyń krwionośnych.
Proces ten jest niezbędny do takich funkcji, jak rozwój embrionalny, gojenie się ran, regeneracja tkanek i tworzenie nowotworów. Celując w VEGFR, można leczyć różne choroby złośliwe i niezłośliwe.
Badacze byli zaintrygowani faktem, iż dwaj członkowie rodziny VEGFR 1 i VEGFR 2 zachowywali się zupełnie inaczej. Podczas gdy VEGFR 2, główny receptor regulujący proces tworzenia nowych naczyń krwionośnych, mógł być spontanicznie aktywowany bez swojego ligandu, drugi członek rodziny VEGFR 1 nie może być spontanicznie aktywowany choćby przy nadmiernej ekspresji w komórkach. Kamufluje się jako martwy enzym VEGFR1 i wiąże się z dziesięciokrotnie większym powinowactwem do swojego ligandu VEGF-A niż VEGFR2. To wiązanie ligandu indukuje przejściową aktywację kinazy (przyspieszającej reakcje chemiczne w organizmie przez enzym).
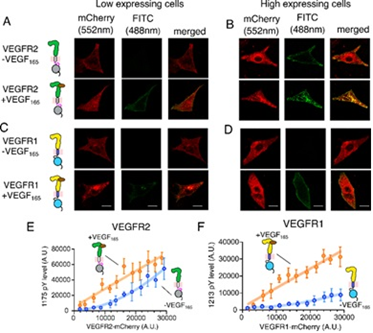
Rysunek 1: Badanie niezależnej od liganda aktywacji VEGFR. A – D Konfokalne obrazy VEGFR2 lub VEGFR1 połączonego z mCherry w liniach komórkowych CHO o niskiej (A, C) i wysokiej (b, d) ekspresji. Poziom ekspresji VEGFR zaznaczono na czerwono, a stan fosforylacji na zielono, pasek skali = 10 µm. Poziom ekspresji VEGFR2 (panel E) lub VEGFR1 (panel F) wykreślono w funkcji poziomu fosforylacji odpowiednich reszt tyrozyny na C-końcowym ogonie
Stwierdzono, iż aktywacja VEGFR1 prowadzi do bólu związanego z nowotworem, przeżycia komórek nowotworowych w przypadku raka piersi i migracji ludzkich komórek raka jelita grubego.
Badając, dlaczego jeden członek rodziny jest tak spontanicznie aktywowany, a drugi ulega autoinhibicji, dr Rahul Das i jego zespół z IISER Kalkuta odkryli, iż unikalny zatrzask jonowy, obecny tylko w VEGFR1, utrzymuje autoinhibicję kinazy w stanie podstawowym. Zatrzask jonowy zaczepia segment przybłonowy o domenę kinazy i stabilizuje autoinhibitowaną konformację VEGFR1.
Badając mechanizm stanu autoinhibicji VEGFR1, badacze zaproponowali kluczową rolę komórkowej fosfatazy tyrozynowej w modulowaniu aktywności VEGFR1. Badania przeprowadzone w Zakładzie Biologii Analitycznej w IISER Kalkuta przy użyciu ITC wspomaganego przez DST-FIST i fluorymetru o zatrzymanym przepływie, podkreśliły potencjał terapeutyczny modulatorów fosfatazy w regulacji patologicznego tworzenia nowych naczyń krwionośnych za pośrednictwem VEGFR1 (angiogenezy), które ma miejsce w rak.
Odkrycie to zostało opublikowane w czasopiśmie Komunikacja przyrodnicza może otworzyć nowe możliwości opracowania interwencji terapeutycznych przeciwko stanom patologicznym w wyniku spontanicznej aktywacji sygnalizacji VEGFR. Małe cząsteczki ukierunkowane na stan autoinhibicji będą miały większy potencjał w leczeniu nowotworów, takich jak ludzki rak jelita grubego i rak nerek, w których występuje nadekspresja VEGFR1.
Link do artykułu:

Rysunek 2: Wiązanie liganda z domeną zewnątrzkomórkową (ECD) indukuje dimeryzację receptora i rearanżację segmentu TM-JM. Powolne uwalnianie hamowania JM w VEGFR1 prowadzi do przejściowej fosforylacji tyrozyny na C-końcowym ogonie. Szybsze uwalnianie hamowania JM w mutantach VEGFR2 lub VEGFR1 przebudowuje fosforylację tyrozyny, aby została podtrzymana. Po lewej: Niezależna od liganda aktywacja VEGFR1 jest tłumiona z powodu delikatnej równowagi pomiędzy powolnym uwalnianiem hamowania JM a aktywnością białkowej fosfatazy tyrozynowej (PTP).

















